Lunesta: Insomnia Medication Treatment (Full Prescribing Information)
Brand Name: Lunesta
Generic Name: Eszopiclone
Dosage Form: tablet, coated
Contents:
Description
Pharmacology
Clinical Trails
Indications and Usage
Contraindications
Warnings
Precautions
Adverse Reactions
Drug Abuse and Dependence
Overdosage
Dosage and Administration
How Supplied
Lunesta patient information (in plain English)
Description
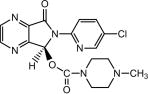
Lunesta (eszopiclone) is a nonbenzodiazepine hypnotic agent that is a pyrrolopyrazine derivative of the cyclopyrrolone class. The chemical name of eszopiclone is (+)-(5S)-6-(5-chloropyridin-2-yl)-7-oxo-6,7-dihydro-5H-pyrrolo[3,4-b] pyrazin-5-yl 4-methylpiperazine-1-carboxylate. Its molecular weight is 388.81, and its empirical formula is C17H17ClN6O3. Eszopiclone has a single chiral center with an (S)-configuration. It has the following chemical structure:
Eszopiclone is a white to light-yellow crystalline solid. Eszopiclone is very slightly soluble in water, slightly soluble in ethanol, and soluble in phosphate buffer (pH 3.2).
Eszopiclone is formulated as film-coated tablets for oral administration. Lunesta tablets contain 1 mg, 2 mg, or 3 mg eszopiclone and the following inactive ingredients: calcium phosphate, colloidal silicon dioxide, croscarmellose sodium, hypromellose, lactose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, titanium dioxide, and triacetin. In addition, both the 1 mg and 3 mg tablets contain FD&C Blue #2.
Clinical Pharmacology
Pharmacodynamics
The precise mechanism of action of eszopiclone as a hypnotic is unknown, but its effect is believed to result from its interaction with GABA-receptor complexes at binding domains located close to or allosterically coupled to benzodiazepine receptors. Eszopiclone is a nonbenzodiazepine hypnotic that is a pyrrolopyrazine derivative of the cyclopyrrolone class with a chemical structure unrelated to pyrazolopyrimidines, imidazopyridines, benzodiazepines, barbiturates, or other drugs with known hypnotic properties.
Pharmacokinetics
The pharmacokinetics of eszopiclone have been investigated in healthy subjects (adult and elderly) and in patients with hepatic disease or renal disease. In healthy subjects, the pharmacokinetic profile was examined after single doses of up to 7.5 mg and after once-daily administration of 1, 3, and 6 mg for 7 days. Eszopiclone is rapidly absorbed, with a time to peak concentration (tmax) of approximately 1 hour and a terminal-phase elimination half-life (t1/2) of approximately 6 hours. In healthy adults, Lunesta does not accumulate with once-daily administration, and its exposure is dose-proportional over the range of 1 to 6 mg.
Absorption And Distribution
Eszopiclone is rapidly absorbed following oral administration. Peak plasma concentrations are achieved within approximately 1 hour after oral administration. Eszopiclone is weakly bound to plasma protein (52-59%). The large free fraction suggests that eszopiclone disposition should not be affected by drug-drug interactions caused by protein binding. The blood-to-plasma ratio for eszopiclone is less than one, indicating no selective uptake by red blood cells.
Metabolism
Following oral administration, eszopiclone is extensively metabolized by oxidation and demethylation. The primary plasma metabolites are (S)-zopiclone-N-oxide and (S)-N-desmethyl zopiclone; the latter compound binds to GABA receptors with substantially lower potency than eszopiclone, and the former compound shows no significant binding to this receptor. In vitro studies have shown that CYP3A4 and CYP2E1 enzymes are involved in the metabolism of eszopiclone. Eszopiclone did not show any inhibitory potential on CYP450 1A2, 2A6, 2C9, 2C19, 2D6, 2E1, and 3A4 in cryopreserved human hepatocytes.
Elimination
After oral administration, eszopiclone is eliminated with a mean t1/2 of approximately 6 hours. Up to 75% of an oral dose of racemic zopiclone is excreted in the urine, primarily as metabolites. A similar excretion profile would be expected for eszopiclone, the S-isomer of racemic zopiclone. Less than 10% of the orally administered eszopiclone dose is excreted in the urine as parent drug.
Effect Of Food
In healthy adults, administration of a 3 mg dose of eszopiclone after a high-fat meal resulted in no change in AUC, a reduction in mean Cmax of 21%, and delayed tmax by approximately 1 hour. The half-life remained unchanged, approximately 6 hours. The effects of Lunesta on sleep onset may be reduced if it is taken with or immediately after a high-fat/heavy meal.
Special Populations
Age
Compared with non-elderly adults, subjects 65 years and older had an increase of 41% in total exposure (AUC) and a slightly prolonged elimination of eszopiclone (t1/2 approximately 9 hours). Cmax was unchanged. Therefore, in elderly patients the starting dose of Lunesta should be decreased to 1 mg and the dose should not exceed 2 mg.
Gender
The pharmacokinetics of eszopiclone in men and women are similar.
Race
In an analysis of data on all subjects participating in Phase 1 studies of eszopiclone, the pharmacokinetics for all races studied appeared similar.
Hepatic Impairment
Pharmacokinetics of a 2 mg eszopiclone dose were assessed in 16 healthy volunteers and in 8 subjects with mild, moderate, and severe liver disease. Exposure was increased 2-fold in severely impaired patients compared with the healthy volunteers. Cmax and tmax were unchanged. The dose of Lunesta should not be increased above 2 mg in patients with severe hepatic impairment. No dose adjustment is necessary for patients with mild-to-moderate hepatic impairment. Lunesta should be used with caution in patients with hepatic impairment. (See DOSAGE AND ADMINISTRATION.)
Renal Impairment
The pharmacokinetics of eszopiclone were studied in 24 patients with mild, moderate, or severe renal impairment. AUC and Cmax were similar in the patients compared with demographically matched healthy control subjects. No dose adjustment is necessary in patients with renal impairment, since less than 10% of the orally administered eszopiclone dose is excreted in the urine as parent drug.
Drug Interactions
Eszopiclone is metabolized by CYP3A4 and CYP2E1 via demethylation and oxidation. There were no pharmacokinetic or pharmacodynamic interactions between eszopiclone and paroxetine, digoxin, or warfarin. When eszopiclone was coadministered with olanzapine, no pharmacokinetic interaction was detected in levels of eszopiclone or olanzapine, but a pharmacodynamic interaction was seen on a measure of psychomotor function. Eszopiclone and lorazepam decreased each other's Cmax by 22%. Coadministration of eszopiclone 3 mg to subjects receiving ketoconazole 400 mg, a potent inhibitor of CYP3A4, resulted in a 2.2-fold increase in exposure to eszopiclone. Lunesta would not be expected to alter the clearance of drugs metabolized by common CYP450 enzymes. (See PRECAUTIONS.)
Clinical Trails
The effect of Lunesta on reducing sleep latency and improving sleep maintenance was established in studies with 2100 subjects (ages 18-86) with chronic and transient insomnia in six placebo-controlled trials of up to 6 months' duration. Two of these trials were in elderly patients (n=523). Overall, at the recommended adult dose (2-3 mg) and elderly dose (1-2 mg), Lunesta significantly decreased sleep latency and improved measures of sleep maintenance (objectively measured as wake time after sleep onset [WASO] and subjectively measured as total sleep time).
Transient Insomnia
Healthy adults were evaluated in a model of transient insomnia (n=436) in a sleep laboratory in a double-blind, parallel-group, single-night trial comparing two doses of eszopiclone and placebo. Lunesta 3 mg was superior to placebo on measures of sleep latency and sleep maintenance, including polysomnographic (PSG) parameters of latency to persistent sleep (LPS) and WASO.
Chronic Insomnia (Adults And Elderly)
The effectiveness of Lunesta was established in five controlled studies in chronic insomnia. Three controlled studies were in adult subjects, and two controlled studies were in elderly subjects with chronic insomnia.
Adults
In the first study, adults with chronic insomnia (n=308) were evaluated in a double-blind, parallel-group trial of 6 weeks' duration comparing Lunesta 2 mg and 3 mg with placebo. Objective endpoints were measured for 4 weeks. Both 2 mg and 3 mg were superior to placebo on LPS at 4 weeks. The 3 mg dose was superior to placebo on WASO.
In the second study, adults with chronic insomnia (n=788) were evaluated using subjective measures in a double-blind, parallel-group trial comparing the safety and efficacy of Lunesta 3 mg with placebo administered nightly for 6 months. Lunesta was superior to placebo on subjective measures of sleep latency, total sleep time, and WASO.
In addition, a 6-period cross-over PSG study evaluating eszopiclone doses of 1 to 3 mg, each given over a 2-day period, demonstrated effectiveness of all doses on LPS, and of 3 mg on WASO. In this trial, the response was dose-related.
Elderly
Elderly subjects (ages 65-86) with chronic insomnia were evaluated in two double-blind, parallel-group trials of 2 weeks' duration. One study (n=231) compared the effects of Lunesta with placebo on subjective outcome measures, and the other (n=292) on objective and subjective outcome measures. The first study compared 1 mg and 2 mg of Lunesta with placebo, while the second study compared 2 mg of Lunesta with placebo. All doses were superior to placebo on measures of sleep latency. In both studies, 2 mg of Lunesta was superior to placebo on measures of sleep maintenance.
Studies Pertinent To Safety Concerns For Sedative/Hypnotic Drugs
Cognitive, Memory, Sedative, and Psychomotor Effects
In two double-blind, placebo-controlled, single-dose cross-over studies of 12 patients each (one study in patients with insomnia; one in normal volunteers), the effects of Lunesta 2 and 3 mg were assessed on 20 measures of cognitive function and memory at 9.5 and 12 hours after a nighttime dose. Although results suggested that patients receiving Lunesta 3 mg performed more poorly than patients receiving placebo on a very small number of these measures at 9.5 hours post-dose, no consistent pattern of abnormalities was seen.
In a 6-month double-blind, placebo-controlled trial of nightly administered Lunesta 3 mg, 8/593 subjects treated with Lunesta 3 mg (1.3%) and 0/195 subjects treated with placebo (0%) spontaneously reported memory impairment. The majority of these events were mild in nature (5/8), and none were reported as severe. Four of these events occurred within the first 7 days of treatment and did not recur. The incidence of spontaneously reported confusion in this 6-month study was 0.5% in both treatment arms. In a 6-week adult study of nightly administered Lunesta 2 mg or 3 mg or placebo, the spontaneous reporting rates for confusion were 0%, 3.0%, and 0%, respectively, and for memory impairment were 1%, 1%, and 0%, respectively.
In a 2-week study of 264 elderly insomniacs randomized to either nightly Lunesta 2 mg or placebo, spontaneous reporting rates of confusion and memory impairment were 0% vs. 0.8% and 1.5% vs. 0%, respectively. In another 2-week study of 231 elderly insomniacs, the spontaneous reporting rates for the 1 mg, 2 mg, and placebo groups for confusion were 0%, 2.5%, and 0%, respectively, and for memory impairment were 1.4%, 0%, and 0%, respectively.
A study of normal subjects exposed to single fixed doses of Lunesta from 1 to 7.5 mg using the DSST to assess sedation and psychomotor function at fixed times after dosing (hourly up to 16 hours) found the expected sedation and reduction in psychomotor function. This was maximal at 1 hour and present up to 4 hours, but was no longer present by 5 hours.
In another study, patients with insomnia were given 2 or 3 mg doses of Lunesta nightly, with DSST assessed on the mornings following days 1, 15, and 29 of treatment. While both the placebo and Lunesta 3 mg groups showed an improvement in DSST scores relative to baseline the following morning (presumably due to a learning effect), the improvement in the placebo group was greater and reached statistical significance on night 1, although not on nights 15 and 29. For the Lunesta 2 mg group, DSST change scores were not significantly different from placebo at any time point.
Withdrawal-Emergent Anxiety And Insomnia
During nightly use for an extended period, pharmacodynamic tolerance or adaptation has been observed with other hypnotics. If a drug has a short elimination half-life, it is possible that a relative deficiency of the drug or its active metabolites (i.e., in relationship to the receptor site) may occur at some point in the interval between each night's use. This is believed to be responsible for two clinical findings reported to occur after several weeks of nightly use of other rapidly eliminated hypnotics: increased wakefulness during the last quarter of the night and the appearance of increased signs of daytime anxiety.
In a 6-month double-blind, placebo-controlled study of nightly administration of Lunesta 3 mg, rates of anxiety reported as an adverse event were 2.1% in the placebo arm and 3.7% in the Lunesta arm. In a 6-week adult study of nightly administration, anxiety was reported as an adverse event in 0%, 2.9%, and 1.0% of the placebo, 2 mg, and 3 mg treatment arms, respectively. In this study, single-blind placebo was administered on nights 45 and 46, the first and second days of withdrawal from study drug. New adverse events were recorded during the withdrawal period, beginning with day 45, up to 14 days after discontinuation. During this withdrawal period, 105 subjects previously taking nightly Lunesta 3 mg for 44 nights spontaneously reported anxiety (1%), abnormal dreams (1.9%), hyperesthesia (1%), and neurosis (1%), while none of 99 subjects previously taking placebo reported any of these adverse events during the withdrawal period.
Rebound insomnia, defined as a dose-dependent temporary worsening in sleep parameters (latency, sleep efficiency, and number of awakenings) compared with baseline following discontinuation of treatment, is observed with short- and intermediate-acting hypnotics. Rebound insomnia following discontinuation of Lunesta relative to placebo and baseline was examined objectively in a 6-week adult study on the first 2 nights of discontinuation (nights 45 and 46) following 44 nights of active treatment with 2 mg or 3 mg. In the Lunesta 2 mg group, compared with baseline, there was a significant increase in WASO and a decrease in sleep efficiency, both occurring only on the first night after discontinuation of treatment. No changes from baseline were noted in the Lunesta 3 mg group on the first night after discontinuation, and there was a significant improvement in LPS and sleep efficiency compared with baseline following the second night of discontinuation. Comparisons of changes from baseline between Lunesta and placebo were also performed. On the first night after discontinuation of Lunesta 2 mg, LPS and WASO were significantly increased and sleep efficiency was reduced; there were no significant differences on the second night. On the first night following discontinuation of Lunesta 3 mg, sleep efficiency was significantly reduced. No other differences from placebo were noted in any other sleep parameter on either the first or second night following discontinuation. For both doses, the discontinuation-emergent effect was mild, had the characteristics of the return of the symptoms of chronic insomnia, and appeared to resolve by the second night after Lunesta discontinuation.
Indications and Usage
Lunesta is indicated for the treatment of insomnia. In controlled outpatient and sleep laboratory studies, Lunesta administered at bedtime decreased sleep latency and improved sleep maintenance.
The clinical trials performed in support of efficacy were up to 6 months in duration. The final formal assessments of sleep latency and maintenance were performed at 4 weeks in the 6-week study (adults only), at the end of both 2-week studies (elderly only) and at the end of the 6-month study (adults only).
Contraindications
None known.
Warnings
Because sleep disturbances may be the presenting manifestation of a physical and/or psychiatric disorder, symptomatic treatment of insomnia should be initiated only after a careful evaluation of the patient. The failure of insomnia to remit after 7 to 10 days of treatment may indicate the presence of a primary psychiatric and/or medical illness that should be evaluated. Worsening of insomnia or the emergence of new thinking or behavior abnormalities may be the consequence of an unrecognized psychiatric or physical disorder. Such findings have emerged during the course of treatment with sedative/hypnotic drugs, including Lunesta. Because some of the important adverse effects of Lunesta appear to be dose-related, it is important to use the lowest possible effective dose, especially in the elderly (see Dosage and Administration).
A variety of abnormal thinking and behavior changes have been reported to occur in association with the use of sedative/hypnotics. Some of these changes may be characterized by decreased inhibition (e.g., aggressiveness and extroversion that seem out of character), similar to effects produced by alcohol and other CNS depressants. Other reported behavioral changes have included bizarre behavior, agitation, hallucinations, and depersonalization. Complex behaviors such as "sleep-driving" (i.e., driving while not fully awake after ingestion of a sedative-hypnotic, with amnesia for the event) have been reported. These events can occur in sedative-hypnotic-naïve as well as in sedative-hypnotic-experienced persons. Although behaviors such as sleep-driving may occur with Lunesta alone at therapeutic doses, the use of alcohol and other CNS depressants with Lunesta appears to increase the risk of such behaviors, as does the use of Lunesta at doses exceeding the maximum recommended dose. Due to the risk to the patient and the community, discontinuation of Lunesta should be strongly considered for patients who report a "sleep-driving" episode. Other complex behaviors (e.g., preparing and eating food, making phone calls, or having sex) have been reported in patients who are not fully awake after taking a sedative-hypnotic. As with sleep-driving, patients usually do not remember these events. Amnesia and other neuropsychiatric symptoms may occur unpredictably. In primarily depressed patients, worsening of depression, including suicidal thoughts and actions (including completed suicides), have been reported in association with the use of sedative/hypnotics.
It can rarely be determined with certainty whether a particular instance of the abnormal behaviors listed above are drug-induced, spontaneous in origin, or a result of an underlying psychiatric or physical disorder. Nonetheless, the emergence of any new behavioral sign or symptom of concern requires careful and immediate evaluation.
Following rapid dose decrease or abrupt discontinuation of the use of sedative/hypnotics, there have been reports of signs and symptoms similar to those associated with withdrawal from other CNS-depressant drugs (see Drug Abuse and Dependence).
Lunesta, like other hypnotics, has CNS-depressant effects. Because of the rapid onset of action, Lunesta should only be ingested immediately prior to going to bed or after the patient has gone to bed and has experienced difficulty falling asleep. Patients receiving Lunesta should be cautioned against engaging in hazardous occupations requiring complete mental alertness or motor coordination (e.g., operating machinery or driving a motor vehicle) after ingesting the drug, and be cautioned about potential impairment of the performance of such activities on the day following ingestion of Lunesta. Lunesta, like other hypnotics, may produce additive CNS-depressant effects when coadministered with other psychotropic medications, anticonvulsants, antihistamines, ethanol, and other drugs that themselves produce CNS depression. Lunesta should not be taken with alcohol. Dose adjustment may be necessary when Lunesta is administered with other CNS-depressant agents, because of the potentially additive effects.
Severe anaphylactic and anaphylactoid reactions
Rare cases of angioedema involving the tongue, glottis or larynx have been reported in patients after taking the first or subsequent doses of sedative-hypnotics, including Lunesta. Some patients have had additional symptoms such as dyspnea, throat closing, or nausea and vomiting that suggest anaphylaxis. Some patients have required medical therapy in the emergency department. If angioedema involves the tongue, glottis or larynx, airway obstruction may occur and be fatal. Patients who develop angioedema after treatment with Lunesta should not be rechallenged with the drug.
Precautions
General
Timing Of Drug Administration
Lunesta should be taken immediately before bedtime. Taking a sedative/hypnotic while still up and about may result in short-term memory impairment, hallucinations, impaired coordination, dizziness, and lightheadedness.
Use In The Elderly And/Or Debilitated Patients
Impaired motor and/or cognitive performance after repeated exposure or unusual sensitivity to sedative/hypnotic drugs is a concern in the treatment of elderly and/or debilitated patients. The recommended starting dose of Lunesta for these patients is 1 mg. (See Dosage and Administration.)
Use In Patients With Concomitant Illness
Clinical experience with eszopiclone in patients with concomitant illness is limited. Eszopiclone should be used with caution in patients with diseases or conditions that could affect metabolism or hemodynamic responses.
A study in healthy volunteers did not reveal respiratory-depressant effects at doses 2.5-fold higher (7 mg) than the recommended dose of eszopiclone. Caution is advised, however, if Lunesta is prescribed to patients with compromised respiratory function.
The dose of Lunesta should be reduced to 1 mg in patients with severe hepatic impairment, because systemic exposure is doubled in such subjects. No dose adjustment appears necessary for subjects with mild or moderate hepatic impairment. No dose adjustment appears necessary in subjects with any degree of renal impairment, since less than 10% of eszopiclone is excreted unchanged in the urine.
The dose of Lunesta should be reduced in patients who are administered potent inhibitors of CYP3A4, such as ketoconazole, while taking Lunesta. Downward dose adjustment is also recommended when Lunesta is administered with agents having known CNS-depressant effects.
Use In Patients With Depression
Sedative/hypnotic drugs should be administered with caution to patients exhibiting signs and symptoms of depression. Suicidal tendencies may be present in such patients, and protective measures may be required. Intentional overdose is more common in this group of patients; therefore, the least amount of drug that is feasible should be prescribed for the patient at any one time.
Information For Patients
Patients should be instructed to read the accompanying Medication Guide with each new prescription and refill. The complete text of the Medication Guide is reprinted at the end of this document. Patients should be given the following information:
Patients should be instructed to take Lunesta immediately prior to going to bed, and only if they can dedicate 8 hours to sleep.
Patients should be instructed not to take Lunesta with alcohol or with other sedating medications.
Patients should be advised to consult with their physician if they have a history of depression, mental illness, or suicidal thoughts, have a history of drug or alcohol abuse, or have liver disease.
Women should be advised to contact their physician if they become pregnant, plan to become pregnant, or if they are nursing.
SPECIAL CONCERNS "Sleep-Driving" and other complex behaviors
There have been reports of people getting out of bed after taking a sedative-hypnotic and driving their cars while not fully awake, often with no memory of the event. If a patient experiences such an episode, it should be reported to his or her doctor immediately, since "sleep-driving" can be dangerous. This behavior is more likely to occur when Lunesta is taken with alcohol or other central nervous system depressants (see Warnings). Other complex behaviors (e.g., preparing and eating food, making phone calls, or having sex) have been reported in patients who are not fully awake after taking a sedative-hypnotic. As with sleep-driving, patients usually do not remember these events.
Laboratory Tests
There are no specific laboratory tests recommended.
Drug Interactions
CNS-Active Drugs
Ethanol: An additive effect on psychomotor performance was seen with coadministration of eszopiclone and ethanol 0.70 g/kg for up to 4 hours after ethanol administration.
Paroxetine: Coadministration of single doses of eszopiclone 3 mg and paroxetine 20 mg daily for 7 days produced no pharmacokinetic or pharmacodynamic interaction.
Lorazepam: Coadministration of single doses of eszopiclone 3 mg and lorazepam 2 mg did not have clinically relevant effects on the pharmacodynamics or pharmacokinetics of either drug.
Olanzapine: Coadministration of eszopiclone 3 mg and olanzapine 10 mg produced a decrease in DSST scores. The interaction was pharmacodynamic; there was no alteration in the pharmacokinetics of either drug.
Drugs That Inhibit CYP3A4 (Ketoconazole)
CYP3A4 is a major metabolic pathway for elimination of eszopiclone. The AUC of eszopiclone was increased 2.2-fold by coadministration of ketoconazole, a potent inhibitor of CYP3A4, 400 mg daily for 5 days. Cmax and t1/2 were increased 1.4-fold and 1.3-fold, respectively. Other strong inhibitors of CYP3A4 (e.g., itraconazole, clarithromycin, nefazodone, troleandomycin, ritonavir, nelfinavir) would be expected to behave similarly.
Drugs That Induce CYP3A4 (Rifampicin)
Racemic zopiclone exposure was decreased 80% by concomitant use of rifampicin, a potent inducer of CYP3A4. A similar effect would be expected with eszopiclone.
Drugs Highly Bound To Plasma Protein
Eszopiclone is not highly bound to plasma proteins (52-59% bound); therefore, the disposition of eszopiclone is not expected to be sensitive to alterations in protein binding. Administration of eszopiclone 3 mg to a patient taking another drug that is highly protein-bound would not be expected to cause an alteration in the free concentration of either drug.
Drugs With A Narrow Therapeutic Index
Digoxin: A single dose of eszopiclone 3 mg did not affect the pharmacokinetics of digoxin measured at steady state following dosing of 0.5 mg twice daily for one day and 0.25 mg daily for the next 6 days.
Warfarin: Eszopiclone 3 mg administered daily for 5 days did not affect the pharmacokinetics of (R)- or (S)-warfarin, nor were there any changes in the pharmacodynamic profile (prothrombin time) following a single 25 mg oral dose of warfarin.
Carcinogenesis, Mutagenesis, Impairment Of Fertility
Carcinogenesis
In a carcinogenicity study in Sprague-Dawley rats in which eszopiclone was given by oral gavage, no increases in tumors were seen; plasma levels (AUC) of eszopiclone at the highest dose used in this study (16 mg/kg/day) are estimated to be 80 (females) and 20 (males) times those in humans receiving the maximum recommended human dose (MRHD). However, in a carcinogenicity study in Sprague-Dawley rats in which racemic zopiclone was given in the diet, and in which plasma levels of eszopiclone were reached that were greater than those reached in the above study of eszopiclone, an increase in mammary gland adenocarcinomas in females and an increase in thyroid gland follicular cell adenomas and carcinomas in males were seen at the highest dose of 100 mg/kg/day. Plasma levels of eszopiclone at this dose are estimated to be 150 (females) and 70 (males) times those in humans receiving the MRHD. The mechanism for the increase in mammary adenocarcinomas is unknown. The increase in thyroid tumors is thought to be due to increased levels of TSH secondary to increased metabolism of circulating thyroid hormones, a mechanism that is not considered to be relevant to humans.
In a carcinogenicity study in B6C3F1 mice in which racemic zopiclone was given in the diet, an increase in pulmonary carcinomas and carcinomas plus adenomas in females and an increase in skin fibromas and sarcomas in males were seen at the highest dose of 100 mg/kg/day. Plasma levels of eszopiclone at this dose are estimated to be 8 (females) and 20 (males) times those in humans receiving the MRHD. The skin tumors were due to skin lesions induced by aggressive behavior, a mechanism that is not relevant to humans. A carcinogenicity study was also performed in which CD-1 mice were given eszopiclone at doses up to 100 mg/kg/day by oral gavage; although this study did not reach a maximum tolerated dose, and was thus inadequate for overall assessment of carcinogenic potential, no increases in either pulmonary or skin tumors were seen at doses producing plasma levels of eszopiclone estimated to be 90 times those in humans receiving the MRHD — i.e., 12 times the exposure in the racemate study.
Eszopiclone did not increase tumors in a p53 transgenic mouse bioassay at oral doses up to 300 mg/kg/day.
Mutagenesis
Eszopiclone was positive in the mouse lymphoma chromosomal aberration assay and produced an equivocal response in the Chinese hamster ovary cell chromosomal aberration assay. It was not mutagenic or clastogenic in the bacterial Ames gene mutation assay, in an unscheduled DNA synthesis assay, or in an in vivo mouse bone marrow micronucleus assay.
(S)-N-desmethyl zopiclone, a metabolite of eszopiclone, was positive in the Chinese hamster ovary cell and human lymphocyte chromosomal aberration assays. It was negative in the bacterial Ames mutation assay, in an in vitro32P-postlabeling DNA adduct assay, and in an in vivo mouse bone marrow chromosomal aberration and micronucleus assay.
Impairment Of Fertility
Eszopiclone was given by oral gavage to male rats at doses up to 45 mg/kg/day from 4 weeks premating through mating and to female rats at doses up to 180 mg/kg/day from 2 weeks premating through day 7 of pregnancy. An additional study was performed in which only females were treated, up to 180 mg/kg/day. Eszopiclone decreased fertility, probably because of effects in both males and females, with no females becoming pregnant when both males and females were treated with the highest dose; the no-effect dose in both sexes was 5 mg/kg (16 times the MRHD on a mg/m2 basis). Other effects included increased pre-implantation loss (no-effect dose 25 mg/kg), abnormal estrus cycles (no-effect dose 25 mg/kg), and decreases in sperm number and motility and increases in morphologically abnormal sperm (no-effect dose 5 mg/kg).
Pregnancy
Pregnancy Category C
Eszopiclone administered by oral gavage to pregnant rats and rabbits during the period of organogenesis showed no evidence of teratogenicity up to the highest doses tested (250 and 16 mg/kg/day in rats and rabbits, respectively; these doses are 800 and 100 times, respectively, the maximum recommended human dose [MRHD] on a mg/m2 basis). In the rat, slight reductions in fetal weight and evidence of developmental delay were seen at maternally toxic doses of 125 and 150 mg/kg/day, but not at 62.5 mg/kg/day (200 times the MRHD on a mg/m2 basis).
Eszopiclone was also administered by oral gavage to pregnant rats throughout the pregnancy and lactation periods at doses of up to 180 mg/kg/day. Increased post-implantation loss, decreased postnatal pup weights and survival, and increased pup startle response were seen at all doses; the lowest dose tested, 60 mg/kg/day, is 200 times the MRHD on a mg/m2 basis. These doses did not produce significant maternal toxicity. Eszopiclone had no effects on other behavioral measures or reproductive function in the offspring.
There are no adequate and well-controlled studies of eszopiclone in pregnant women. Eszopiclone should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Labor And Delivery
Lunesta has no established use in labor and delivery.
Nursing Mothers
It is not known whether Lunesta is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when Lunesta is administered to a nursing woman.
Pediatric Use
Safety and effectiveness of eszopiclone in children below the age of 18 have not been established.
Geriatric Use
A total of 287 subjects in double-blind, parallel-group, placebo-controlled clinical trials who received eszopiclone were 65 to 86 years of age. The overall pattern of adverse events for elderly subjects (median age = 71 years) in 2-week studies with nighttime dosing of 2 mg eszopiclone was not different from that seen in younger adults (see Adverse Reactions, Table 2). Lunesta 2 mg exhibited significant reduction in sleep latency and improvement in sleep maintenance in the elderly population.
Adverse Reactions
The premarketing development program for Lunesta included eszopiclone exposures in patients and/or normal subjects from two different groups of studies: approximately 400 normal subjects in clinical pharmacology/pharmacokinetic studies, and approximately 1550 patients in placebo-controlled clinical effectiveness studies, corresponding to approximately 263 patient-exposure years. The conditions and duration of treatment with Lunesta varied greatly and included (in overlapping categories) open-label and double-blind phases of studies, inpatients and outpatients, and short-term and longer-term exposure. Adverse reactions were assessed by collecting adverse events, results of physical examinations, vital signs, weights, laboratory analyses, and ECGs.
Adverse events during exposure were obtained primarily by general inquiry and recorded by clinical investigators using terminology of their own choosing. Consequently, it is not possible to provide a meaningful estimate of the proportion of individuals experiencing adverse events without first grouping similar types of events into a smaller number of standardized event categories. In the tables and tabulations that follow, COSTART terminology has been used to classify reported adverse events.
The stated frequencies of adverse events represent the proportion of individuals who experienced, at least once, a treatment-emergent adverse event of the type listed. An event was considered treatment-emergent if it occurred for the first time or worsened while the patient was receiving therapy following baseline evaluation.
Adverse Findings Observed In Placebo-Controlled Trials
Adverse Events Resulting In Discontinuation Of Treatment
In placebo-controlled, parallel-group clinical trials in the elderly, 3.8% of 208 patients who received placebo, 2.3% of 215 patients who received 2 mg Lunesta, and 1.4% of 72 patients who received 1 mg Lunesta discontinued treatment due to an adverse event. In the 6-week parallel-group study in adults, no patients in the 3 mg arm discontinued because of an adverse event. In the long-term 6-month study in adult insomnia patients, 7.2% of 195 patients who received placebo and 12.8% of 593 patients who received 3 mg Lunesta discontinued due to an adverse event. No event that resulted in discontinuation occurred at a rate of greater than 2%.
Adverse Events Observed At An Incidence Of ≥2% In Controlled Trials
Table 1 shows the incidence of treatment-emergent adverse events from a Phase 3 placebo-controlled study of Lunesta at doses of 2 or 3 mg in non-elderly adults. Treatment duration in this trial was 44 days. The table includes only events that occurred in 2% or more of patients treated with Lunesta 2 mg or 3 mg in which the incidence in patients treated with Lunesta was greater than the incidence in placebo-treated patients.
| Adverse Event | Placebo (n=99) |
Lunesta 2 mg (n=104) |
Lunesta 3 mg (n=105) |
|---|---|---|---|
| 1 Events for which the Lunesta incidence was equal to or less than placebo are not listed on the table, but included the following: abnormal dreams, accidental injury, back pain, diarrhea, flu syndrome, myalgia, pain, pharyngitis, and rhinitis. | |||
| * Gender-specific adverse event in females | |||
| ** Gender-specific adverse event in males | |||
| Body as a Whole | |||
| Headache | 13 | 21 | 17 |
| Viral Infection | 1 | 3 | 3 |
| Digestive System | |||
| Dry Mouth | 3 | 5 | 7 |
| Dyspepsia | 4 | 4 | 5 |
| Nausea | 4 | 5 | 4 |
| Vomiting | 1 | 3 | 0 |
| Nervous System | |||
| Anxiety | 0 | 3 | 1 |
| Confusion | 0 | 0 | 3 |
| Depression | 0 | 4 | 1 |
| Dizziness | 4 | 5 | 7 |
| Hallucinations | 0 | 1 | 3 |
| Libido Decreased | 0 | 0 | 3 |
| Nervousness | 3 | 5 | 0 |
| Somnolence | 3 | 10 | 8 |
| Respiratory System | |||
| Infection | 3 | 5 | 10 |
| Skin and Appendages | |||
| Rash | 1 | 3 | 4 |
| Special Senses | |||
| Unpleasant Taste | 3 | 17 | 34 |
| Urogenital System | |||
| Dysmenorrhea * | 0 | 3 | 0 |
| Gynecomastia ** | 0 | 3 | 0 |
Adverse events from Table 1 that suggest a dose-response relationship in adults include viral infection, dry mouth, dizziness, hallucinations, infection, rash, and unpleasant taste, with this relationship clearest for unpleasant taste.
Table 2 shows the incidence of treatment-emergent adverse events from combined Phase 3 placebo-controlled studies of Lunesta at doses of 1 or 2 mg in elderly adults (ages 65-86). Treatment duration in these trials was 14 days. The table includes only events that occurred in 2% or more of patients treated with Lunesta 1 mg or 2 mg in which the incidence in patients treated with Lunesta was greater than the incidence in placebo-treated patients.
| Adverse Event | Placebo (n=208) |
Lunesta 1 mg (n=72) |
Lunesta 2 mg (n=215) |
|---|---|---|---|
| 1 Events for which the Lunesta incidence was equal to or less than placebo are not listed on the table, but included the following: abdominal pain, asthenia, nausea, rash, and somnolence. | |||
| Body as a Whole | |||
| Accidental Injury | 1 | 0 | 3 |
| Headache | 14 | 15 | 13 |
| Pain | 2 | 4 | 5 |
| Digestive System | |||
| Diarrhea | 2 | 4 | 2 |
| Dry Mouth | 2 | 3 | 7 |
| Dyspepsia | 2 | 6 | 2 |
| Nervous System | |||
| Abnormal Dreams | 0 | 3 | 1 |
| Dizziness | 2 | 1 | 6 |
| Nervousness | 1 | 0 | 2 |
| Neuralgia | 0 | 3 | 0 |
| Skin and Appendages | |||
| Pruritus | 1 | 4 | 1 |
| Special Senses | |||
| Unpleasant Taste | 0 | 8 | 12 |
| Urogenital System | |||
| Urinary Tract Infection | 0 | 3 | 0 |
Adverse events from Table 2 that suggest a dose-response relationship in elderly adults include pain, dry mouth, and unpleasant taste, with this relationship again clearest for unpleasant taste.
These figures cannot be used to predict the incidence of adverse events in the course of usual medical practice because patient characteristics and other factors may differ from those that prevailed in the clinical trials. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different treatments, uses, and investigators. The cited figures, however, do provide the prescribing physician with some basis for estimating the relative contributions of drug and non-drug factors to the adverse event incidence rate in the population studied.
Other Events Observed During The Premarketing Evaluation Of Lunesta
Following is a list of modified COSTART terms that reflect treatment-emergent adverse events as defined in the introduction to the ADVERSE REACTIONS section and reported by approximately 1550 subjects treated with Lunesta at doses in the range of 1 to 3.5 mg/day during Phase 2 and 3 clinical trials throughout the United States and Canada. All reported events are included except those already listed in Tables 1 and 2 or elsewhere in labeling, minor events common in the general population, and events unlikely to be drug-related. Although the events reported occurred during treatment with Lunesta, they were not necessarily caused by it.
Events are further categorized by body system and listed in order of decreasing frequency according to the following definitions: frequent adverse events are those that occurred on one or more occasions in at least 1/100 patients; infrequent adverse events are those that occurred in fewer than 1/100 patients but in at least 1/1,000 patients; rare adverse events are those that occurred in fewer than 1/1,000 patients. Gender-specific events are categorized based on their incidence for the appropriate gender.
Body as a Whole: Frequent: chest pain; Infrequent: allergic reaction, cellulitis, face edema, fever, halitosis, heat stroke, hernia, malaise, neck rigidity, photosensitivity.
Cardiovascular System: Frequent: migraine; Infrequent: hypertension; Rare: thrombophlebitis.
Digestive System: Infrequent: anorexia, cholelithiasis, increased appetite, melena, mouth ulceration, thirst, ulcerative stomatitis; Rare: colitis, dysphagia, gastritis, hepatitis, hepatomegaly, liver damage, stomach ulcer, stomatitis, tongue edema, rectal hemorrhage.
Hemic and Lymphatic System: Infrequent: anemia, lymphadenopathy.
Metabolic and Nutritional: Frequent: peripheral edema; Infrequent: hypercholesteremia, weight gain, weight loss; Rare: dehydration, gout, hyperlipemia, hypokalemia.
Musculoskeletal System: Infrequent: arthritis, bursitis, joint disorder (mainly swelling, stiffness, and pain), leg cramps, myasthenia, twitching; Rare: arthrosis, myopathy, ptosis.
Nervous System: Infrequent: agitation, apathy, ataxia, emotional lability, hostility, hypertonia, hypesthesia, incoordination, insomnia, memory impairment, neurosis, nystagmus, paresthesia, reflexes decreased, thinking abnormal (mainly difficulty concentrating), vertigo; Rare: abnormal gait, euphoria, hyperesthesia, hypokinesia, neuritis, neuropathy, stupor, tremor.
Respiratory System: Infrequent: asthma, bronchitis, dyspnea, epistaxis, hiccup, laryngitis.
Skin and Appendages: Infrequent: acne, alopecia, contact dermatitis, dry skin, eczema, skin discoloration, sweating, urticaria; Rare: erythema multiforme, furunculosis, herpes zoster, hirsutism, maculopapular rash, vesiculobullous rash.
Special Senses: Infrequent: conjunctivitis, dry eyes, ear pain, otitis externa, otitis media, tinnitus, vestibular disorder; Rare: hyperacusis, iritis, mydriasis, photophobia.
Urogenital System: Infrequent: amenorrhea, breast engorgement, breast enlargement, breast neoplasm, breast pain, cystitis, dysuria, female lactation, hematuria, kidney calculus, kidney pain, mastitis, menorrhagia, metrorrhagia, urinary frequency, urinary incontinence, uterine hemorrhage, vaginal hemorrhage, vaginitis; Rare: oliguria, pyelonephritis, urethritis.
Drug Abuse and Dependence:
Controlled Substance Class
Lunesta is a Schedule IV controlled substance under the Controlled Substances Act. Other substances under the same classification are benzodiazepines and the nonbenzodiazepine hypnotics zaleplon and zolpidem. While eszopiclone is a hypnotic agent with a chemical structure unrelated to benzodiazepines, it shares some of the pharmacologic properties of the benzodiazepines.
Abuse, Dependence, And Tolerance
Abuse And Dependence
Abuse and addiction are separate and distinct from physical dependence and tolerance. Abuse is characterized by misuse of the drug for non-medical purposes, often in combination with other psychoactive substances. Physical dependence is a state of adaptation that is manifested by a specific withdrawal syndrome that can be produced by abrupt cessation, rapid dose reduction, decreasing blood level of the drug and/or administration of an antagonist. Tolerance is a state of adaptation in which exposure to a drug induces changes that result in a diminution of one or more of the drug's effects over time. Tolerance may occur to both the desired and undesired effects of drugs and may develop at different rates for different effects.
Addiction is a primary, chronic, neurobiological disease with genetic, psychosocial, and environmental factors influencing its development and manifestations. It is characterized by behaviors that include one or more of the following: impaired control over drug use, compulsive use, continued use despite harm, and craving. Drug addiction is a treatable disease, utilizing a multidisciplinary approach, but relapse is common.
In a study of abuse liability conducted in individuals with known histories of benzodiazepine abuse, eszopiclone at doses of 6 and 12 mg produced euphoric effects similar to those of diazepam 20 mg. In this study, at doses 2-fold or greater than the maximum recommended doses, a dose-related increase in reports of amnesia and hallucinations was observed for both Lunesta and diazepam.
The clinical trial experience with Lunesta revealed no evidence of a serious withdrawal syndrome. Nevertheless, the following adverse events included in DSM-IV criteria for uncomplicated sedative/hypnotic withdrawal were reported during clinical trials following placebo substitution occurring within 48 hours following the last Lunesta treatment: anxiety, abnormal dreams, nausea, and upset stomach. These reported adverse events occurred at an incidence of 2% or less. Use of benzodiazepines and similar agents may lead to physical and psychological dependence. The risk of abuse and dependence increases with the dose and duration of treatment and concomitant use of other psychoactive drugs. The risk is also greater for patients who have a history of alcohol or drug abuse or history of psychiatric disorders. These patients should be under careful surveillance when receiving Lunesta or any other hypnotic.
Tolerance
Some loss of efficacy to the hypnotic effect of benzodiazepines and benzodiazepine-like agents may develop after repeated use of these drugs for a few weeks.
No development of tolerance to any parameter of sleep measurement was observed over six months. Tolerance to the efficacy of Lunesta 3 mg was assessed by 4-week objective and 6-week subjective measurements of time to sleep onset and sleep maintenance for Lunesta in a placebo-controlled 44-day study, and by subjective assessments of time to sleep onset and WASO in a placebo-controlled study for 6 months.
Overdosage
There is limited premarketing clinical experience with the effects of an overdosage of Lunesta. In clinical trials with eszopiclone, one case of overdose with up to 36 mg of eszopiclone was reported in which the subject fully recovered. Individuals have fully recovered from racemic zopiclone overdoses up to 340 mg (56 times the maximum recommended dose of eszopiclone).
Signs And Symptoms
Signs and symptoms of overdose effects of CNS depressants can be expected to present as exaggerations of the pharmacological effects noted in preclinical testing. Impairment of consciousness ranging from somnolence to coma has been described. Rare individual instances of fatal outcomes following overdose with racemic zopiclone have been reported in European postmarketing reports, most often associated with overdose with other CNS-depressant agents.
Recommended Treatment
General symptomatic and supportive measures should be used along with immediate gastric lavage where appropriate. Intravenous fluids should be administered as needed. Flumazenil may be useful. As in all cases of drug overdose, respiration, pulse, blood pressure, and other appropriate signs should be monitored and general supportive measures employed. Hypotension and CNS depression should be monitored and treated by appropriate medical intervention. The value of dialysis in the treatment of overdosage has not been determined.
Poison Control Center
As with the management of all overdosage, the possibility of multiple drug ingestion should be considered. The physician may wish to consider contacting a poison control center for up-to-date information on the management of hypnotic drug product overdosage.
Dosage and Administration
The dose of Lunesta should be individualized. The recommended starting dose for Lunesta for most non-elderly adults is 2 mg immediately before bedtime. Dosing can be initiated at or raised to 3 mg if clinically indicated, since 3 mg is more effective for sleep maintenance (see PRECAUTIONS).
The recommended starting dose of Lunesta for elderly patients whose primary complaint is difficulty falling asleep is 1 mg immediately before bedtime. In these patients, the dose may be increased to 2 mg if clinically indicated. For elderly patients whose primary complaint is difficulty staying asleep, the recommended dose is 2 mg immediately before bedtime (see Precautions).
Taking Lunesta with or immediately after a heavy, high-fat meal results in slower absorption and would be expected to reduce the effect of Lunesta on sleep latency (see Pharmacokinetics under Clinical Pharmacology).
Special Populations
Hepatic
The starting dose of Lunesta should be 1 mg in patients with severe hepatic impairment. Lunesta should be used with caution in these patients.
Coadministration With CYP3A4 Inhibitors
The starting dose of Lunesta should not exceed 1 mg in patients coadministered Lunesta with potent CYP3A4 inhibitors. If needed, the dose can be raised to 2 mg.
How Supplied
Lunesta 3 mg tablets are round, dark blue, film-coated, and identified with debossed markings of S193 on one side.
Lunesta 2 mg tablets are round, white, film-coated, and identified with debossed markings of S191 on one side.
Lunesta 1 mg tablets are round, light blue, film-coated, and identified with debossed markings of S190 on one side.
Store at 25°C (77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
They are supplied as follows:
| NDC | Strength | Color | Bottle Count |
| 0339-4162-11 | 2 mg | WHITE | 90 |
| 0339-4163-06 | 3 mg | BLUE | 30 |
| 0339-4163-11 | 3 mg | BLUE | 90 |
Last Updated: 01/2009
Lunesta patient information (in plain English)
Detailed Info on Signs, Symptoms, Causes, Treatments of Sleep Disorders
The information in this monograph is not intended to cover all possible uses, directions, precautions, drug interactions or adverse effects. This information is generalized and is not intended as specific medical advice. If you have questions about the medicines you are taking or would like more information, check with your doctor, pharmacist, or nurse.
back to:
~ all articles on sleeping disorders
APA Reference
Staff, H.
(2019, August 31). Lunesta: Insomnia Medication Treatment (Full Prescribing Information), HealthyPlace. Retrieved
on 2026, March 28 from https://www.healthyplace.com/other-info/sleep-disorders/lunesta-insomnia-medication-treatment-full-prescribing-information
")