Januvia Diabetes Treatment - Januvia Full Prescribing Information
Brand Name: Januvia
Generic Name: Sitagliptin
Contents:
Indications and Usage
Dosage and Administration
Dosage Forms and Strengths
Contraindications
Warnings and Precautions
Adverse Reactions
Drug Interactions
Use in Specific Populations
Overdose
Description
Pharmacology
Nonclinical Toxicology
Clinical Studies
How Supplied
Januvia, sitagliptin, patient information sheet (in plain English)
Indications and Usage
Monotherapy and Combination Therapy
Januvia is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.[See Clinical Studies.]
Important Limitations of Use
Januvia should not be used in patients with type 1 diabetes or for the treatment of diabetic ketoacidosis, as it would not be effective in these settings.
Januvia has not been studied in combination with insulin.
Dosage and Administration
Recommended Dosing
The recommended dose of Januvia is 100 mg once daily. Januvia can be taken with or without food.
Patients with Renal Insufficiency
For patients with mild renal insufficiency (creatinine clearance [CrCl] greater than or equal to 50 mL/min, approximately corresponding to serum creatinine levels of less than or equal to 1.7 mg/dL in men and less than or equal to 1.5 mg/dL in women), no dosage adjustment for Januvia is required.
For patients with moderate renal insufficiency (CrCl greater than or equal to 30 to less than 50 mL/min, approximately corresponding to serum creatinine levels of greater than 1.7 to less than or equal to 3.0 mg/dL in men and greater than 1.5 to less than or equal to 2.5 mg/dL in women), the dose of Januvia is 50 mg once daily.
For patients with severe renal insufficiency (CrCl less than 30 mL/min, approximately corresponding to serum creatinine levels of greater than 3.0 mg/dL in men and greater than 2.5 mg/dL in women) or with end-stage renal disease (ESRD) requiring hemodialysis or peritoneal dialysis, the dose of Januvia is 25 mg once daily. Januvia may be administered without regard to the timing of hemodialysis.
Because there is a need for dosage adjustment based upon renal function, assessment of renal function is recommended prior to initiation of Januvia and periodically thereafter. Creatinine clearance can be estimated from serum creatinine using the Cockcroft-Gault formula. [See Clinical Pharmacology.]
Concomitant Use with a Sulfonylurea
When Januvia is used in combination with a sulfonylurea, a lower dose of sulfonylurea may be required to reduce the risk of hypoglycemia. [See Warnings and Precautions.]
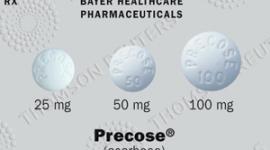
Dosage Forms and Strengths
- 100 mg tablets are beige, round, film-coated tablets with "277" on one side.
- 50 mg tablets are light beige, round, film-coated tablets with "112" on one side.
- 25 mg tablets are pink, round, film-coated tablets with "221" on one side.
Contraindications
History of a serious hypersensitivity reaction to sitagliptin, such as anaphylaxis or angioedema. [See Warnings and Precautions and Adverse Reactions.]
Warnings and Precautions
Use in Patients with Renal Insufficiency
A dosage adjustment is recommended in patients with moderate or severe renal insufficiency and in patients with ESRD requiring hemodialysis or peritoneal dialysis. [See Dosage and Administration; Clinical Pharmacology.]
Use with Medications Known to Cause Hypoglycemia
As is typical with other antihyperglycemic agents used in combination with a sulfonylurea, when Januvia was used in combination with a sulfonylurea, a class of medications known to cause hypoglycemia, the incidence of hypoglycemia was increased over that of placebo. [See Adverse Reactions.] Therefore, a lower dose of sulfonylurea may be required to reduce the risk of hypoglycemia. [See Dosage and Administration.]
Hypersensitivity Reactions
There have been postmarketing reports of serious hypersensitivity reactions in patients treated with Januvia. These reactions include anaphylaxis, angioedema, and exfoliative skin conditions including Stevens-Johnson syndrome. Because these reactions are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure. Onset of these reactions occurred within the first 3 months after initiation of treatment with Januvia, with some reports occurring after the first dose. If a hypersensitivity reaction is suspected, discontinue Januvia, assess for other potential causes for the event, and institute alternative treatment for diabetes. [See Adverse Reactions.]
Macrovascular Outcomes
There have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with Januvia or any other anti-diabetic drug.
Adverse Reactions
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In controlled clinical studies as both monotherapy and combination therapy with metformin or pioglitazone, the overall incidence of adverse reactions, hypoglycemia, and discontinuation of therapy due to clinical adverse reactions with Januvia were similar to placebo. In combination with glimepiride, with or without metformin, the overall incidence of clinical adverse reactions with Januvia was higher than with placebo, in part related to a higher incidence of hypoglycemia (see Table 1); the incidence of discontinuation due to clinical adverse reactions was similar to placebo.
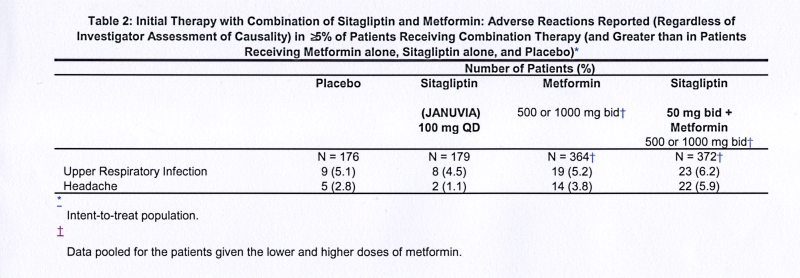
Two placebo-controlled monotherapy studies, one of 18- and one of 24-week duration, included patients treated with Januvia 100 mg daily, Januvia 200 mg daily, and placebo. Three 24-week, placebo-controlled add-on combination therapy studies, one with metformin, one with pioglitazone, and one with glimepiride with or without metformin, were also conducted. In addition to a stable dose of metformin, pioglitazone, glimepiride, or glimepiride and metformin, patients whose diabetes was not adequately controlled were given either Januvia 100 mg daily or placebo. The adverse reactions, reported regardless of investigator assessment of causality in ≥5% of patients treated with Januvia 100 mg daily as monotherapy, Januvia in combination with pioglitazone, or Januvia in combination with glimepiride, with or without metformin, and more commonly than in patients treated with placebo, are shown in Table 1.
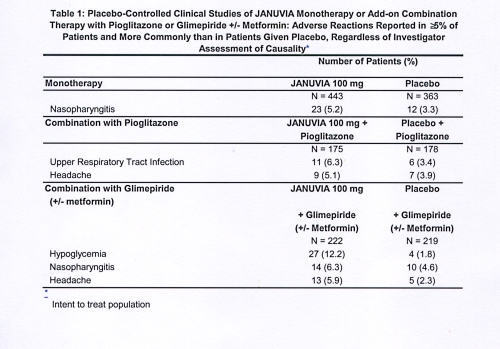
In the study of patients receiving Januvia as add-on combination therapy with metformin, there were no adverse reactions reported regardless of investigator assessment of causality in ≥5% of patients and more commonly than in patients given placebo.
In the prespecified pooled analysis of the two monotherapy studies, the add-on to metformin study, and the add-on to pioglitazone study, the overall incidence of adverse reactions of hypoglycemia in patients treated with Januvia 100 mg was similar to placebo (1.2% vs 0.9%). Adverse reactions of hypoglycemia were based on all reports of hypoglycemia; a concurrent glucose measurement was not required. The incidence of selected gastrointestinal adverse reactions in patients treated with Januvia was as follows: abdominal pain (Januvia 100 mg, 2.3%; placebo, 2.1%), nausea (1.4%, 0.6%), and diarrhea (3.0%, 2.3%).
In an additional, 24-week, placebo-controlled factorial study of initial therapy with sitagliptin in combination with metformin, the adverse reactions reported (regardless of investigator assessment of causality) in ≥5% of patients are shown in Table 2. The incidence of hypoglycemia was 0.6% in patients given placebo, 0.6% in patients given sitagliptin alone, 0.8% in patients given metformin alone, and 1.6% in patients given sitagliptin in combination with metformin.
No clinically meaningful changes in vital signs or in ECG (including in QTc interval) were observed in patients treated with Januvia.
Laboratory Tests
Across clinical studies, the incidence of laboratory adverse reactions was similar in patients treated with Januvia 100 mg compared to patients treated with placebo. A small increase in white blood cell count (WBC) was observed due to an increase in neutrophils. This increase in WBC (of approximately 200 cells/microL vs placebo, in four pooled placebo-controlled clinical studies, with a mean baseline WBC count of approximately 6600 cells/microL) is not considered to be clinically relevant. In a 12-week study of 91 patients with chronic renal insufficiency, 37 patients with moderate renal insufficiency were randomized to Januvia 50 mg daily, while 14 patients with the same magnitude of renal impairment were randomized to placebo. Mean (SE) increases in serum creatinine were observed in patients treated with Januvia [0.12 mg/dL (0.04)] and in patients treated with placebo [0.07 mg/dL (0.07)]. The clinical significance of this added increase in serum creatinine relative to placebo is not known.
Postmarketing Experience
The following additional adverse reactions have been identified during postapproval use of Januvia. Because these reactions are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hypersensitivity reactions include anaphylaxis, angioedema, rash, urticaria, cutaneous vasculitis, and exfoliative skin conditions including Stevens-Johnson syndrome [see Warnings and Precautions]; hepatic enzyme elevations; pancreatitis.
Drug Interactions
Digoxin
There was a slight increase in the area under the curve (AUC, 11%) and mean peak drug concentration (Cmax, 18%) of digoxin with the co-administration of 100 mg sitagliptin for 10 days. Patients receiving digoxin should be monitored appropriately. No dosage adjustment of digoxin or Januvia is recommended.
Use in Specific Populations
Pregnancy
Pregnancy Category B:
Reproduction studies have been performed in rats and rabbits. Doses of sitagliptin up to 125 mg/kg (approximately 12 times the human exposure at the maximum recommended human dose) did not impair fertility or harm the fetus. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed. Merck & Co., Inc. maintains a registry to monitor the pregnancy outcomes of women exposed to Januvia while pregnant. Health care providers are encouraged to report any prenatal exposure to Januvia by calling the Pregnancy Registry at (800) 986-8999.
Sitagliptin administered to pregnant female rats and rabbits from gestation day 6 to 20 (organogenesis) was not teratogenic at oral doses up to 250 mg/kg (rats) and 125 mg/kg (rabbits), or approximately 30- and 20-times human exposure at the maximum recommended human dose (MRHD) of 100 mg/day based on AUC comparisons. Higher doses increased the incidence of rib malformations in offspring at 1000 mg/kg, or approximately 100 times human exposure at the MRHD.
Sitagliptin administered to female rats from gestation day 6 to lactation day 21 decreased body weight in male and female offspring at 1000 mg/kg. No functional or behavioral toxicity was observed in offspring of rats.
Placental transfer of sitagliptin administered to pregnant rats was approximately 45% at 2 hours and 80% at 24 hours postdose. Placental transfer of sitagliptin administered to pregnant rabbits was approximately 66% at 2 hours and 30% at 24 hours.
Nursing Mothers
Sitagliptin is secreted in the milk of lactating rats at a milk to plasma ratio of 4:1. It is not known whether sitagliptin is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when Januvia is administered to a nursing woman.
Pediatric Use
Safety and effectiveness of Januvia in pediatric patients under 18 years of age have not been established.
Geriatric Use
Of the total number of subjects (N=3884) in pre-approval clinical safety and efficacy studies of Januvia, 725 patients were 65 years and over, while 61 patients were 75 years and over. No overall differences in safety or effectiveness were observed between subjects 65 years and over and younger subjects. While this and other reported clinical experience have not identified differences in responses between the elderly and younger patients, greater sensitivity of some older individuals cannot be ruled out.
This drug is known to be substantially excreted by the kidney. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection in the elderly, and it may be useful to assess renal function in these patients prior to initiating dosing and periodically thereafter [see Dosage and Administration; Clinical Pharmacology].
Overdose
During controlled clinical trials in healthy subjects, single doses of up to 800 mg Januvia were administered. Maximal mean increases in QTc of 8.0 msec were observed in one study at a dose of 800 mg Januvia, a mean effect that is not considered clinically important [see Clinical Pharmacology]. There is no experience with doses above 800 mg in humans. In Phase I multiple-dose studies, there were no dose-related clinical adverse reactions observed with Januvia with doses of up to 600 mg per day for periods of up to 10 days and 400 mg per day for up to 28 days.
In the event of an overdose, it is reasonable to employ the usual supportive measures, e.g., remove unabsorbed material from the gastrointestinal tract, employ clinical monitoring (including obtaining an electrocardiogram), and institute supportive therapy as dictated by the patient's clinical status.
Sitagliptin is modestly dialyzable. In clinical studies, approximately 13.5% of the dose was removed over a 3- to 4-hour hemodialysis session. Prolonged hemodialysis may be considered if clinically appropriate. It is not known if sitagliptin is dialyzable by peritoneal dialysis.
Description
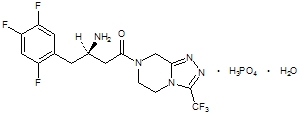
Januvia Tablets contain sitagliptin phosphate, an orally-active inhibitor of the dipeptidyl peptidase-4 (DPP-4) enzyme.
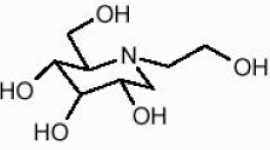
Sitagliptin phosphate monohydrate is described chemically as 7 - [(3R) - 3 - amino - 1 - oxo - 4 - (2,4,5 - trifluorophenyl)butyl] - 5,6,7,8 - tetrahydro - 3 - (trifluoromethyl) - 1,2,4 - triazolo[4,3 - a]pyrazine phosphate (1:1) monohydrate.
The empirical formula is C16H15F6N5O-H3PO4-H2O and the molecular weight is 523.32. The structural formula is:
Sitagliptin phosphate monohydrate is a white to off-white, crystalline, non-hygroscopic powder. It is soluble in water and N,N-dimethyl formamide; slightly soluble in methanol; very slightly soluble in ethanol, acetone, and acetonitrile; and insoluble in isopropanol and isopropyl acetate.
Each film-coated tablet of Januvia contains 32.13, 64.25, or 128.5 mg of sitagliptin phosphate monohydrate, which is equivalent to 25, 50, or 100 mg, respectively, of free base and the following inactive ingredients: microcrystalline cellulose, anhydrous dibasic calcium phosphate, croscarmellose sodium, magnesium stearate, and sodium stearyl fumarate. In addition, the film coating contains the following inactive ingredients: polyvinyl alcohol, polyethylene glycol, talc, titanium dioxide, red iron oxide, and yellow iron oxide.
Clinical Pharmacology
Mechanism Of Action
Sitagliptin is a DPP-4 inhibitor, which is believed to exert its actions in patients with type 2 diabetes by slowing the inactivation of incretin hormones. Concentrations of the active intact hormones are increased by Januvia, thereby increasing and prolonging the action of these hormones. Incretin hormones, including glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), are released by the intestine throughout the day, and levels are increased in response to a meal. These hormones are rapidly inactivated by the enzyme, DPP-4. The incretins are part of an endogenous system involved in the physiologic regulation of glucose homeostasis. When blood glucose concentrations are normal or elevated, GLP-1 and GIP increase insulin synthesis and release from pancreatic beta cells by intracellular signaling pathways involving cyclic AMP. GLP-1 also lowers glucagon secretion from pancreatic alpha cells, leading to reduced hepatic glucose production. By increasing and prolonging active incretin levels, Januvia increases insulin release and decreases glucagon levels in the circulation in a glucose-dependent manner. Sitagliptin demonstrates selectivity for DPP-4 and does not inhibit DPP-8 or DPP-9 activity in vitro at concentrations approximating those from therapeutic doses.
Pharmacodynamics
General
In patients with type 2 diabetes, administration of Januvia led to inhibition of DPP-4 enzyme activity for a 24-hour period. After an oral glucose load or a meal, this DPP-4 inhibition resulted in a 2- to 3-fold increase in circulating levels of active GLP-1 and GIP, decreased glucagon concentrations, and increased responsiveness of insulin release to glucose, resulting in higher C-peptide and insulin concentrations. The rise in insulin with the decrease in glucagon was associated with lower fasting glucose concentrations and reduced glucose excursion following an oral glucose load or a meal.
In a two-day study in healthy subjects, sitagliptin alone increased active GLP-1 concentrations, whereas metformin alone increased active and total GLP-1 concentrations to similar extents. Co-administration of sitagliptin and metformin had an additive effect on active GLP-1 concentrations. Sitagliptin, but not metformin, increased active GIP concentrations. It is unclear how these findings relate to changes in glycemic control in patients with type 2 diabetes.
In studies with healthy subjects, Januvia did not lower blood glucose or cause hypoglycemia.
Cardiac Electrophysiology
In a randomized, placebo-controlled crossover study, 79 healthy subjects were administered a single oral dose of Januvia 100 mg, Januvia 800 mg (8 times the recommended dose), and placebo. At the recommended dose of 100 mg, there was no effect on the QTc interval obtained at the peak plasma concentration, or at any other time during the study. Following the 800 mg dose, the maximum increase in the placebo-corrected mean change in QTc from baseline was observed at 3 hours postdose and was 8.0 msec. This increase is not considered to be clinically significant. At the 800 mg dose, peak sitagliptin plasma concentrations were approximately 11 times higher than the peak concentrations following a 100 mg dose.
In patients with type 2 diabetes administered Januvia 100 mg (N=81) or Januvia 200 mg (N=63) daily, there were no meaningful changes in QTc interval based on ECG data obtained at the time of expected peak plasma concentration.
Pharmacokinetics
The pharmacokinetics of sitagliptin has been extensively characterized in healthy subjects and patients with type 2 diabetes. After oral administration of a 100 mg dose to healthy subjects, sitagliptin was rapidly absorbed, with peak plasma concentrations (median Tmax) occurring 1 to 4 hours postdose. Plas
ma AUC of sitagliptin increased in a dose-proportional manner. Following a single oral 100 mg dose to healthy volunteers, mean plasma AUC of sitagliptin was 8.52 μM-hr, Cmax was 950 nM, and apparent terminal half-life (t1/2) was 12.4 hours. Plasma AUC of sitagliptin increased approximately 14% following 100 mg doses at steady-state compared to the first dose. The intra-subject and inter-subject coefficients of variation for sitagliptin AUC were small (5.8% and 15.1%). The pharmacokinetics of sitagliptin was generally similar in healthy subjects and in patients with type 2 diabetes.
Absorption
The absolute bioavailability of sitagliptin is approximately 87%. Because coadministration of a high-fat meal with Januvia had no effect on the pharmacokinetics, Januvia may be administered with or without food.
Distribution
The mean volume of distribution at steady state following a single 100 mg intravenous dose of sitagliptin to healthy subjects is approximately 198 liters. The fraction of sitagliptin reversibly bound to plasma proteins is low (38%).
Metabolism
Approximately 79% of sitagliptin is excreted unchanged in the urine with metabolism being a minor pathway of elimination.
Following a [14C]sitagliptin oral dose, approximately 16% of the radioactivity was excreted as metabolites of sitagliptin. Six metabolites were detected at trace levels and are not expected to contribute to the plasma DPP-4 inhibitory activity of sitagliptin. In vitro studies indicated that the primary enzyme responsible for the limited metabolism of sitagliptin was CYP3A4, with contribution from CYP2C8.
Excretion
Following administration of an oral [14C]sitagliptin dose to healthy subjects, approximately 100% of the administered radioactivity was eliminated in feces (13%) or urine (87%) within one week of dosing. The apparent terminal t1/2 following a 100 mg oral dose of sitagliptin was approximately 12.4 hours and renal clearance was approximately 350 mL/min.
Elimination of sitagliptin occurs primarily via renal excretion and involves active tubular secretion. Sitagliptin is a substrate for human organic anion transporter-3 (hOAT-3), which may be involved in the renal elimination of sitagliptin. The clinical relevance of hOAT-3 in sitagliptin transport has not been established. Sitagliptin is also a substrate of p-glycoprotein, which may also be involved in mediating the renal elimination of sitagliptin. However, cyclosporine, a p-glycoprotein inhibitor, did not reduce the renal clearance of sitagliptin.
Special Populations
Renal Insufficiency
A single-dose, open-label study was conducted to evaluate the pharmacokinetics of Januvia (50 mg dose) in patients with varying degrees of chronic renal insufficiency compared to normal healthy control subjects. The study included patients with renal insufficiency classified on the basis of creatinine clearance as mild (50 to less than 80 mL/min), moderate (30 to less than 50 mL/min), and severe (less than 30 mL/min), as well as patients with ESRD on hemodialysis. In addition, the effects of renal insufficiency on sitagliptin pharmacokinetics in patients with type 2 diabetes and mild or moderate renal insufficiency were assessed using population pharmacokinetic analyses. Creatinine clearance was measured by 24‑hour urinary creatinine clearance measurements or estimated from serum creatinine based on the Cockcroft‑Gault formula:
CrCl = [140 - age (years)] x weight (kg)
[72 x serum creatinine (mg/dL)]
Compared to normal healthy control subjects, an approximate 1.1- to 1.6-fold increase in plasma AUC of sitagliptin was observed in patients with mild renal insufficiency. Because increases of this magnitude are not clinically relevant, dosage adjustment in patients with mild renal insufficiency is not necessary. Plasma AUC levels of sitagliptin were increased approximately 2-fold and 4-fold in patients with moderate renal insufficiency and in patients with severe renal insufficiency, including patients with ESRD on hemodialysis, respectively. Sitagliptin was modestly removed by hemodialysis (13.5% over a 3- to 4-hour hemodialysis session starting 4 hours postdose). To achieve plasma concentrations of sitagliptin similar to those in patients with normal renal function, lower dosages are recommended in patients with moderate and severe renal insufficiency, as well as in ESRD patients requiring hemodialysis. [See Dosage and Administration (2.2).]
Hepatic Insufficiency
In patients with moderate hepatic insufficiency (Child-Pugh score 7 to 9), mean AUC and Cmax of sitagliptin increased approximately 21% and 13%, respectively, compared to healthy matched controls following administration of a single 100 mg dose of Januvia. These differences are not considered to be clinically meaningful. No dosage adjustment for Januvia is necessary for patients with mild or moderate hepatic insufficiency.
There is no clinical experience in patients with severe hepatic insufficiency (Child-Pugh score >9).
Body Mass Index (BMI)
No dosage adjustment is necessary based on BMI. Body mass index had no clinically meaningful effect on the pharmacokinetics of sitagliptin based on a composite analysis of Phase I pharmacokinetic data and on a population pharmacokinetic analysis of Phase I and Phase II data.
Gender
No dosage adjustment is necessary based on gender. Gender had no clinically meaningful effect on the pharmacokinetics of sitagliptin based on a composite analysis of Phase I pharmacokinetic data and on a population pharmacokinetic analysis of Phase I and Phase II data.
Geriatric
No dosage adjustment is required based solely on age. When the effects of age on renal function are taken into account, age alone did not have a clinically meaningful impact on the pharmacokinetics of sitagliptin based on a population pharmacokinetic analysis. Elderly subjects (65 to 80 years) had approximately 19% higher plasma concentrations of sitagliptin compared to younger subjects.
Pediatric
Studies characterizing the pharmacokinetics of sitagliptin in pediatric patients have not been performed.
Race
No dosage adjustment is necessary based on race. Race had no clinically meaningful effect on the pharmacokinetics of sitagliptin based on a composite analysis of available pharmacokinetic data, including subjects of white, Hispanic, black, Asian, and other racial groups.
Drug Interactions
In Vitro Assessment of Drug Interactions
Sitagliptin is not an inhibitor of CYP isozymes CYP3A4, 2C8, 2C9, 2D6, 1A2, 2C19 or 2B6, and is not an inducer of CYP3A4. Sitagliptin is a p‑glycoprotein substrate, but does not inhibit p‑glycoprotein mediated transport of digoxin. Based on these results, sitagliptin is considered unlikely to cause interactions with other drugs that utilize these pathways.
Sitagliptin is not extensively bound to plasma proteins. Therefore, the propensity of sitagliptin to be involved in clinically meaningful drug‑drug interactions mediated by plasma protein binding displacement is very low.
In Vivo Assessment of Drug Interactions
Effects of Sitagliptin on Other Drugs
In clinical studies, as described below, sitagliptin did not meaningfully alter the pharmacokinetics of metformin, glyburide, simvastatin, rosiglitazone, warfarin, or oral contraceptives, providing in vivo evidence of a low propensity for causing drug interactions with substrates of CYP3A4, CYP2C8, CYP2C9, and organic cationic transporter (OCT).
Digoxin: Sitagliptin had a minimal effect on the pharmacokinetics of digoxin. Following administration of 0.25 mg digoxin concomitantly with 100 mg of Januvia daily for 10 days, the plasma AUC of digoxin was increased by 11%, and the plasma Cmax by 18%.
Metformin: Co-administration of multiple twice-daily doses of sitagliptin with metformin, an OCT substrate, did not meaningfully alter the pharmacokinetics of metformin in patients with type 2 diabetes. Therefore, sitagliptin is not an inhibitor of OCT-mediated transport.
Sulfonylureas: Single-dose pharmacokinetics of glyburide, a CYP2C9 substrate, was not meaningfully altered in subjects receiving multiple doses of sitagliptin. Clinically meaningful interactions would not be expected with other sulfonylureas (e.g., glipizide, tolbutamide, and glimepiride) which, like glyburide, are primarily eliminated by CYP2C9.
Simvastatin: Single-dose pharmacokinetics of simvastatin, a CYP3A4 substrate, was not meaningfully altered in subjects receiving multiple daily doses of sitagliptin. Therefore, sitagliptin is not an inhibitor of CYP3A4-mediated metabolism.
Thiazolidinediones: Single-dose pharmacokinetics of rosiglitazone, was not meaningfully altered in subjects receiving multiple daily doses of sitagliptin, indicating that Januvia is not an inhibitor of CYP2C8-mediated metabolism.
Warfarin: Multiple daily doses of sitagliptin did not meaningfully alter the pharmacokinetics, as assessed by measurement of S(-) or R(+) warfarin enantiomers, or pharmacodynamics (as assessed by measurement of prothrombin INR) of a single dose of warfarin. Because S(-) warfarin is primarily metabolized by CYP2C9, these data also support the conclusion that sitagliptin is not a CYP2C9 inhibitor.
Oral Contraceptives: Co-administration with sitagliptin did not meaningfully alter the steady-state pharmacokinetics of norethindrone or ethinyl estradiol.
Effects of Other Drugs on Sitagliptin
Clinical data described below suggest that sitagliptin is not susceptible to clinically meaningful interactions by co-administered medications.
Metformin: Co-administration of multiple twice-daily doses of metformin with sitagliptin did not meaningfully alter the pharmacokinetics of sitagliptin in patients with type 2 diabetes.
Cyclosporine: A study was conducted to assess the effect of cyclosporine, a potent inhibitor of p-glycoprotein, on the pharmacokinetics of sitagliptin. Co-administration of a single 100 mg oral dose of Januvia and a single 600 mg oral dose of cyclosporine increased the AUC and Cmax of sitagliptin by approximately 29% and 68%, respectively. These modest changes in sitagliptin pharmacokinetics were not considered to be clinically meaningful. The renal clearance of sitagliptin was also not meaningfully altered. Therefore, meaningful interactions would not be expected with other p-glycoprotein inhibitors.
Nonclinical Toxicology
Carcinogenesis, Mutagenesis, Impairment Of Fertility
A two-year carcinogenicity study was conducted in male and female rats given oral doses of sitagliptin of 50, 150, and 500 mg/kg/day. There was an increased incidence of combined liver adenoma/carcinoma in males and females and of liver carcinoma in females at 500 mg/kg. This dose results in exposures approximately 60 times the human exposure at the maximum recommended daily adult human dose (MRHD) of 100 mg/day based on AUC comparisons. Liver tumors were not observed at 150 mg/kg, approximately 20 times the human exposure at the MRHD. A two-year carcinogenicity study was conducted in male and female mice given oral doses of sitagliptin of 50, 125, 250, and 500 mg/kg/day. There was no increase in the incidence of tumors in any organ up to 500 mg/kg, approximately 70 times human exposure at the MRHD. Sitagliptin was not mutagenic or clastogenic with or without metabolic activation in the Ames bacterial mutagenicity assay, a Chinese hamster ovary (CHO) chromosome aberration assay, an in vitro cytogenetics assay in CHO, an in vitro rat hepatocyte DNA alkaline elution assay, and an in vivo micronucleus assay.
In rat fertility studies with oral gavage doses of 125, 250, and 1000 mg/kg, males were treated for 4 weeks prior to mating, during mating, up to scheduled termination (approximately 8 weeks total) and females were treated 2 weeks prior to mating through gestation day 7. No adverse effect on fertility was observed at 125 mg/kg (approximately 12 times human exposure at the MRHD of 100 mg/day based on AUC comparisons). At higher doses, nondose-related increased resorptions in females were observed (approximately 25 and 100 times human exposure at the MRHD based on AUC comparison).
Clinical Studies
There were approximately 3800 patients with type 2 diabetes randomized in six double-blind, placebo-controlled clinical safety and efficacy studies conducted to evaluate the effects of sitagliptin on glycemic control. The ethnic/racial distribution in these studies was approximately 60% white, 20% Hispanic, 8% Asian, 6% black, and 6% other groups. Patients had an overall mean age of approximately 55 years (range 18 to 87 years). In addition, an active (glipizide)-controlled study of 52-weeks duration was conducted in 1172 patients with type 2 diabetes who had inadequate glycemic control on metformin.
In patients with type 2 diabetes, treatment with Januvia produced clinically significant improvements in hemoglobin A1C, fasting plasma glucose (FPG) and 2-hour post-prandial glucose (PPG) compared to placebo.
Monotherapy
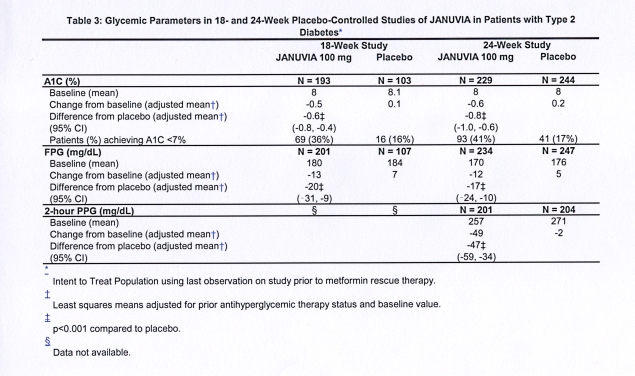
A total of 1262 patients with type 2 diabetes participated in two double-blind, placebo-controlled studies, one of 18-week and another of 24-week duration, to evaluate the efficacy and safety of Januvia monotherapy. In both monotherapy studies, patients currently on an antihyperglycemic agent discontinued the agent, and underwent a diet, exercise, and drug wash-out period of about 7 weeks. Patients with inadequate glycemic control (A1C 7% to 10%) after the wash-out period were randomized after completing a 2-week single-blind placebo run-in period; patients not currently on antihyperglycemic agents (off therapy for at least 8 weeks) with inadequate glycemic control (A1C 7% to 10%) were randomized after completing the 2-week single-blind placebo run-in period. In the 18-week study, 521 patients were randomized to placebo, Januvia 100 mg, or Januvia 200 mg, and in the 24-week study 741 patients were randomized to placebo, Januvia 100 mg, or Januvia 200 mg. Patients who failed to meet specific glycemic goals during the studies were treated with metformin rescue, added on to placebo or Januvia.
Treatment with Januvia at 100 mg daily provided significant improvements in A1C, FPG, and 2-hour PPG compared to placebo (Table 3). In the 18-week study, 9% of patients receiving Januvia 100 mg and 17% who received placebo required rescue therapy. In the 24-week study, 9% of patients receiving Januvia 100 mg and 21% of patients receiving placebo required rescue therapy. The improvement in A1C compared to placebo was not affected by gender, age, race, prior antihyperglycemic therapy, or baseline BMI. As is typical for trials of agents to treat type 2 diabetes, the mean reduction in A1C with Januvia appears to be related to the degree of A1C elevation at baseline. In these 18- and 24-week studies, among patients who were not on an antihyperglycemic agent at study entry, the reductions from baseline in A1C were -0.7% and -0.8%, respectively, for those given Januvia, and -0.1% and -0.2%, respectively, for those given placebo. Overall, the 200 mg daily dose did not provide greater glycemic efficacy than the 100 mg daily dose. The effect of Januvia on lipid endpoints was similar to placebo. Body weight did not increase from baseline with Januvia therapy in either study, compared to a small reduction in patients given placebo.
Additional Monotherapy Study
A multinational, randomized, double-blind, placebo-controlled study was also conducted to assess the safety and tolerability of Januvia in 91 patients with type 2 diabetes and chronic renal insufficiency (creatinine clearance less than 50 mL/min). Patients with moderate renal insufficiency received 50 mg daily of Januvia and those with severe renal insufficiency or with ESRD on hemodialysis or peritoneal dialysis received 25 mg daily. In this study, the safety and tolerability of Januvia were generally similar to placebo. A small increase in serum creatinine was reported in patients with moderate renal insufficiency treated with Januvia relative to those on placebo. In addition, the reductions in A1C and FPG with Januvia compared to placebo were generally similar to those observed in other monotherapy studies. [See Clinical Pharmacology.]
Combination Therapy
Add-on Combination Therapy with Metformin
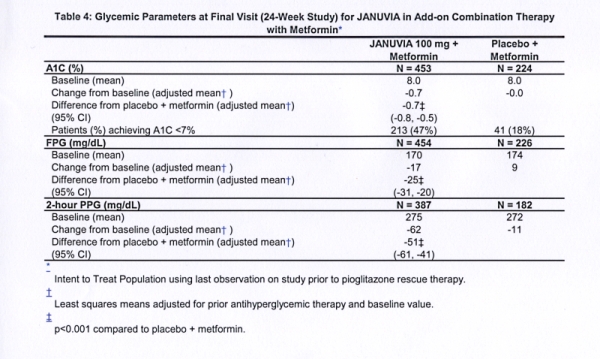
A total of 701 patients with type 2 diabetes participated in a 24-week, randomized, double-blind, placebo-controlled study designed to assess the efficacy of Januvia in combination with metformin. Patients already on metformin (N=431) at a dose of at least 1500 mg per day were randomized after completing a 2-week single-blind placebo run-in period. Patients on metformin and another antihyperglycemic agent (N=229) and patients not on any antihyperglycemic agents (off therapy for at least 8 weeks, N=41) were randomized after a run-in period of approximately 10 weeks on metformin (at a dose of at least 1500 mg per day) in monotherapy. Patients with inadequate glycemic control (A1C 7% to 10%) were randomized to the addition of either 100 mg of Januvia or placebo, administered once daily. Patients who failed to meet specific glycemic goals during the studies were treated with pioglitazone rescue.
In combination with metformin, Januvia provided significant improvements in A1C, FPG, and 2-hour PPG compared to placebo with metformin (Table 4). Rescue glycemic therapy was used in 5% of patients treated with Januvia 100 mg and 14% of patients treated with placebo. A similar decrease in body weight was observed for both treatment groups.
Initial Combination Therapy with Metformin
A total of 1091 patients with type 2 diabetes and inadequate glycemic control on diet and exercise participated in a 24-week, randomized, double-blind, placebo-controlled factorial study designed to assess the efficacy of sitagliptin as initial therapy in combination with metformin. Patients on an antihyperglycemic agent (N=541) discontinued the agent, and underwent a diet, exercise, and drug washout period of up to 12 weeks duration. After the washout period, patients with inadequate glycemic control (A1C 7.5% to 11%) were randomized after completing a 2-week single-blind placebo run-in period. Patients not on antihyperglycemic agents at study entry (N=550) with inadequate glycemic control (A1C 7.5% to 11%) immediately entered the 2-week single-blind placebo run-in period and then were randomized. Approximately equal numbers of patients were randomized to receive initial therapy with placebo, 100 mg of Januvia once daily, 500 mg or 1000 mg of metformin twice daily, or 50 mg of sitagliptin twice daily in combination with 500 mg or 1000 mg of metformin twice daily. Patients who failed to meet specific glycemic goals during the study were treated with glyburide (glibenclamide) rescue.
Initial therapy with the combination of Januvia and metformin provided significant improvements in A1C, FPG, and 2-hour PPG compared to placebo, to metformin alone, and to Januvia alone (Table 5, Figure 1). Mean reductions from baseline in A1C were generally greater for patients with higher baseline A1C values. For patients not on an antihyperglycemic agent at study entry, mean reductions from baseline in A1C were: Januvia 100 mg once daily, -1.1%; metformin 500 mg bid, -1.1%; metformin 1000 mg bid, -1.2%; sitagliptin 50 mg bid with metformin 500 mg bid, -1.6%; sitagliptin 50 mg bid with metformin 1000 mg bid, -1.9%; and for patients receiving placebo, -0.2%. Lipid effects were generally neutral. The decrease in body weight in the groups given sitagliptin in combination with metformin was similar to that in the groups given metformin alone or placebo.
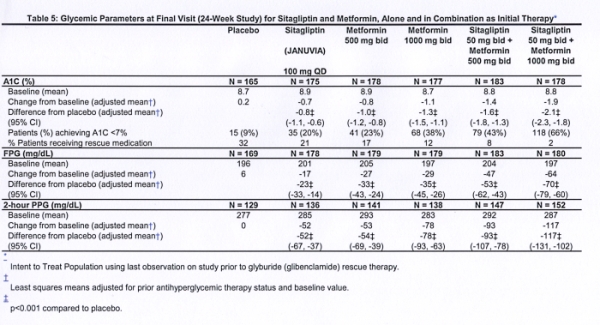
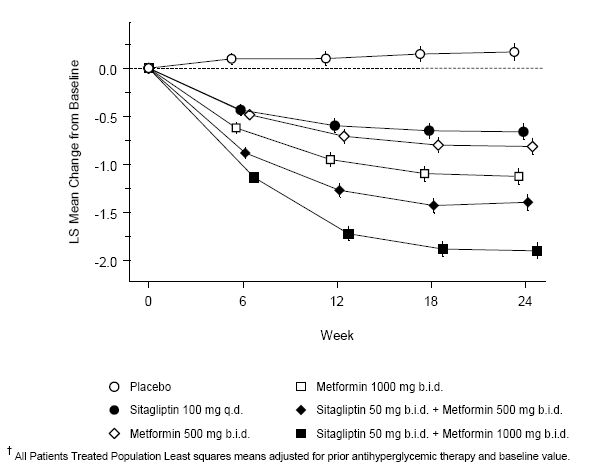
In addition, this study included patients (N=117) with more severe hyperglycemia (A1C greater than 11% or blood glucose greater than 280 mg/dL) who were treated with twice daily open-label Januvia 50 mg and metformin 1000 mg. In this group of patients, the mean baseline A1C value was 11.2%, mean FPG was 314 mg/dL, and mean 2-hour PPG was 441 mg/dL. After 24 weeks, mean decreases from baseline of -2.9% for A1C, -127 mg/dL for FPG, and -208 mg/dL for 2-hour PPG were observed.
Initial combination therapy or maintenance of combination therapy may not be appropriate for all patients. These management options are left to the discretion of the health care provider.
Active-Controlled Study vs Glipizide in Combination with Metformin
The efficacy of Januvia was evaluated in a 52-week, double-blind, glipizide-controlled noninferiority trial in patients with type 2 diabetes. Patients not on treatment or on other antihyperglycemic agents entered a run-in treatment period of up to 12 weeks duration with metformin monotherapy (dose of greater than or equal to 1500 mg per day) which included washout of medications other than metformin, if applicable. After the run-in period, those with inadequate glycemic control (A1C 6.5% to 10%) were randomized 1:1 to the addition of Januvia 100 mg once daily or glipizide for 52 weeks. Patients receiving glipizide were given an initial dosage of 5 mg/day and then electively titrated over the next 18 weeks to a maximum dosage of 20 mg/day as needed to optimize glycemic control. Thereafter, the glipizide dose was to be kept constant, except for down-titration to prevent hypoglycemia. The mean dose of glipizide after the titration period was 10 mg.
After 52 weeks, Januvia and glipizide had similar mean reductions from baseline in A1C in the intent-to-treat analysis (Table 6). These results were consistent with the per protocol analysis (Figure 2). A conclusion in favor of the non-inferiority of Januvia to glipizide may be limited to patients with baseline A1C comparable to those included in the study (over 70% of patients had baseline A1C less than 8% and over 90% had A1C less than 9%).

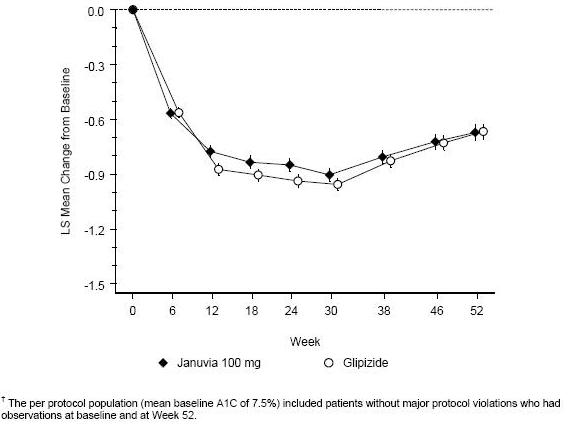
The incidence of hypoglycemia in the Januvia group (4.9%) was significantly (p less than 0.001) lower than that in the glipizide group (32.0%). Patients treated with Januvia exhibited a significant mean decrease from baseline in body weight compared to a significant weight gain in patients administered glipizide (-1.5 kg vs +1.1 kg).
Add-on Combination Therapy with Pioglitazone
A total of 353 patients with type 2 diabetes participated in a 24-week, randomized, double-blind, placebo-controlled study designed to assess the efficacy of Januvia in combination with pioglitazone. Patients on any oral antihyperglycemic agent in monotherapy (N=212) or on a PPARγ agent in combination therapy (N=106) or not on an antihyperglycemic agent (off therapy for at least 8 weeks, N=34) were switched to monotherapy with pioglitazone (at a dose of 30-45 mg per day), and completed a run-in period of approximately 12 weeks in duration. After the run-in period on pioglitazone monotherapy, patients with inadequate glycemic control (A1C 7% to 10%) were randomized to the addition of either 100 mg of Januvia or placebo, administered once daily. Patients who failed to meet specific glycemic goals during the studies were treated with metformin rescue. Glycemic endpoints measured were A1C and fasting glucose.
In combination with pioglitazone, Januvia provided significant improvements in A1C and FPG compared to placebo with pioglitazone (Table 7). Rescue therapy was used in 7% of patients treated with Januvia 100 mg and 14% of patients treated with placebo. There was no significant difference between Januvia and placebo in body weight change.

Add-on Combination Therapy with Glimepiride, with or without Metformin
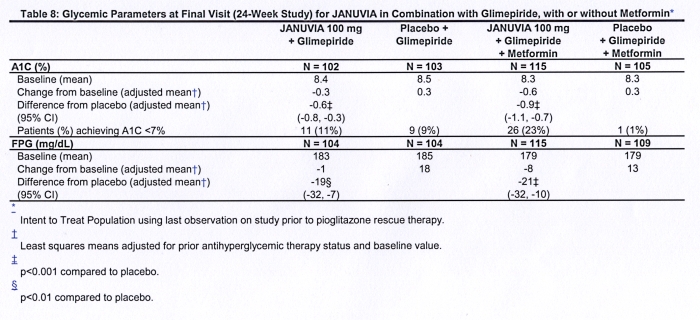
A total of 441 patients with type 2 diabetes participated in a 24-week, randomized, double-blind, placebo-controlled study designed to assess the efficacy of Januvia in combination with glimepiride, with or without metformin. Patients entered a run-in treatment period on glimepiride (greater than or equal to 4 mg per day) alone or glimepiride in combination with metformin (greater than or equal to 1500 mg per day). After a dose-titration and dose-stable run-in period of up to 16 weeks and a 2-week placebo run-in period, patients with inadequate glycemic control (A1C 7.5% to 10.5%) were randomized to the addition of either 100 mg of Januvia or placebo, administered once daily. Patients who failed to meet specific glycemic goals during the studies were treated with pioglitazone rescue.
In combination with glimepiride, with or without metformin, Januvia provided significant improvements in A1C and FPG compared to placebo (Table 8). In the entire study population (patients on Januvia in combination with glimepiride and patients on Januvia in combination with glimepiride and metformin), a mean reduction from baseline relative to placebo in A1C of -0.7% and in FPG of -20 mg/dL was seen. Rescue therapy was used in 12% of patients treated with Januvia 100 mg and 27% of patients treated with placebo. In this study, patients treated with Januvia had a mean increase in body weight of 1.1 kg vs. placebo (+0.8 kg vs. -0.4 kg). In addition, there was an increased rate of hypoglycemia. [See Warnings and Precautions; Adverse Reactions.]
How Supplied
No. 6738 — Tablets Januvia, 50 mg, are light beige, round, film-coated tablets with "112" on one side. They are supplied as follows:
NDC 54868-6031-0 unit-of-use bottles of 30
NDC 54868-6031-1 unit-of-use bottles of 90.
No. 6739 — Tablets Januvia, 100 mg, are beige, round, film-coated tablets with "277" on one side. They are supplied as follows:
NDC 54868-5840-0 unit-of-use bottles of 30.
Storage
Store at 20-25°C (68-77°F), excursions permitted to 15-30°C (59-86°F), [see USP Controlled Room Temperature].
Last Updated: 09/09
Januvia, sitagliptin, patient information sheet (in plain English)
Detailed Info on Signs, Symptoms, Causes, Treatments of Diabetes
The information in this monograph is not intended to cover all possible uses, directions, precautions, drug interactions or adverse effects. This information is generalized and is not intended as specific medical advice. If you have questions about the medicines you are taking or would like more information, check with your doctor, pharmacist, or nurse.
back to: Browse all Medications for Diabetes
APA Reference
Staff, H.
(2009, September 1). Januvia Diabetes Treatment - Januvia Full Prescribing Information, HealthyPlace. Retrieved
on 2026, May 24 from https://www.healthyplace.com/diabetes/medications/januvia-sitagliptin-for-type-2-diabetes